







數字PCR技術進展簡介
聚合酶鏈式反應 ( polymerase chain reaction,PCR) 提出至今已有20年時間,期間PCR已發展成為分子生物學領域的一項關鍵技術和常規技術,極大地推動了生命科學各個領域的發展。特別是 90 年代后期,美國 ABI 公司推出的實時熒光定量PCR( real time PCR,qPCR) 技術及相關產品更是將PCR由體外合成及定性/半定量檢測技術發展成為一種高靈敏、高特異性和精確定量的基因分析技術。
盡管經過十幾年時間的迅速發展,qPCR 技術已經用于除外傷和營養缺乏癥外所有疾病的診斷,但是,在 PCR擴增過程中影響其擴增效率的因素有很多,不能保證在反應過程中擴增效率保持不變和實際樣品與標準樣品以及不同樣品之間的擴增效率是相同的,由此導至其定量分析所依賴的基礎——循環閾值(CT)不是恒定不變的。因此 qPCR 的定量只是“相對定量”,其準確度和重現性依然不能夠滿足分子生物學定量分析的要求。
20 世紀末,Vogelstein等提出數字PCR(digital PCR,dPCR) 的概念,通過將一個樣本分成幾十到幾萬份,分配到不同的反應單元,每個單元包含一個或多個拷貝的目標分子( DNA 模板) ,在每個反應單元中分別對目標分子進行PCR擴增,擴增結束后對各個反應單元的熒光信號進行統計學分析。與 qPCR 不同的是,數字PCR不依賴于CT值,因此不受擴增效率影響,擴增結束后通過直接計數或泊松分布公式來計算每個反應單元的平均濃度(含量),能夠將誤差控制在5%以內,數字 PCR 可以不需要對照標準樣品和標準曲線來實現絕對定量分析。
該技術提出至今雖然只有十幾年時間,但是由于其獨特的技術優勢和應用前景,使得其產業化發展相當迅速。迄今為止,已有包括Fluidigm和 Bio-Rad等幾家公司相繼推出了數字 PCR 產品,并已經應用于單細胞分析、癌癥早期診斷和產前診斷等研究領域。目前,有關數字 PCR 技術的綜述類文獻并不多見,本文將在現有文獻基礎上,對該技術的原理、定量方法、分類及應用進行評述,并對發展趨勢進行展望。
?
數字PCR(也可稱單分子PCR) 一般包括兩部分內容,即PCR擴增和熒光信號分析。在PCR 擴增階段,與傳統技術不同,數字PCR一般需要將樣品稀釋到單分子水平,并平均分配到幾十至幾萬個單元中進行反應。不同于qPCR 對每個循環進行實時熒光測定的方法,數字 PCR 技術是在擴增結束后對每個反應單元的熒光信號進行采集。最后通過直接計數或泊松分布公式計算得到樣品的原始濃度或含量。
最初 Vogelstein等提出的數字 PCR 是在96孔板中進行的,如圖 1 所示。DNA 模板被稀釋成大約平均每兩個孔內有一個拷貝的濃度,在經過優化的實驗條件下進行 PCR 擴增。他們設計了兩種帶有不同熒光基團的分子信標探針分別與 PCR 產物雜交,其中一種探針可以與野生型和突變型兩種產物雜交,另一種探針只與野生型雜交,通過直接計數每個孔內的熒光信號得到同一樣品中等位基因(或野生型與突變型) 的數目和比值,并利用統計學方法分析樣品間的顯著性差異。目前的數字 PCR 技術主要采用分子信標和TaqMan探針兩種方式對 PCR 產物進行熒光標記。其中分子信標法(如圖1b所示) 通過一對通用引物得到包括野生型和突變型 在內的 PCR 產物,再經過不對稱 PCR( asymmetric PCR) 得到單鏈 DNA 分子與兩種熒光分子信標分別雜交,利用熒光顏色區別野生型和突變型,通過具有不同熒光反應單元數量的多少和比率進行分析,這種方法也被稱為數字SNP( digital singlenucleotide polymorphism,digital SNP) 。TaqMan 探針法則可用于基因表達分析和單細胞多重 PCR等。Vogelstein 等提出一種基于磁珠和微乳液的固相數字 PCR 技術——BEAMing ( beads, emulsion,amplification,magnetics) ,原理如圖 1c 所示。BEAMing 技術通過將引物化學鍵合在磁珠表面,再將單個磁珠與目標分子包裹在微乳液滴中進行 PCR 擴增,將野生型和突變型目標分子在磁珠表面進行復制。擴增結束后進行破乳,再利用流式細胞技術進行熒光計數。該技術還通過預擴增反應,提高了系統的靈敏度,適合用于低概率的等位基因突變分析。BEAMing 技術可通過固液分離除去多余熒光探針,降低背景干擾,可采用普通熒光探針代替高成本分子信標和 TaqMan 探針,降低成本。但是該方法需要將單個目標分子與單個磁珠包裹在同一液滴中,增加了操作的復雜性和難度,需要進行大量條件優化實驗。此外, Zhong 等提出通過調節熒光探針濃度實現多重PCR的技術,實現了SMA基因拷貝數變異和 c.815A>G突變位點等五重PCR分析,突破了通常只有4個熒光通道的局限。
?
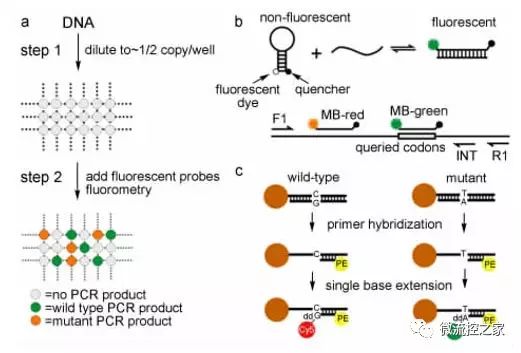
圖 1 數字 PCR 技術原理: a) 數字 PCR 過程,第一步稀釋樣本分配至每個反應單元進行 PCR 反應,第二步熒光檢測;b) 分子信標基因突變分析原理;c) BEAMing 檢測原理
?
傳統的qPCR通常以循環閾值( cycle threshold,CT)為定量分析的基礎,認為在指數擴增的開始階段樣品間的細小誤差尚未放大且擴增效率恒定。對于一個 PCR 反應,到達循環閾值時
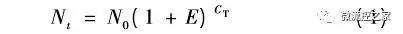
其中,N0 為初始模板的拷貝數,Nt 為第CT個循環時產物的拷貝數,E為擴增效率。將上式兩邊取對數,得到
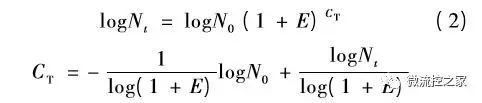
?
對于一個特定的PCR反應,擴增效率E和CT個循環時的拷貝數Nt均為定值,因此,CT值與初始模板拷貝數N0的對數成反比關系。然而,在PCR 擴增過程當中影響其擴增效率的因素有很多,比如酶和引物濃度等,因此很難保證擴增效率不變,導至定量PCR結果的準確度和精密度難以保證。
與傳統qPCR方法不同的是數字PCR采用直接計數的方法進行定量分析,也就是在PCR擴增結束后有熒光信號(產物) 記為 1,無熒光信號(產物) 記為 0,有熒光信號的反應單元中至少包含一個拷貝的目標分子。理論上,在樣品中的目標DNA濃度極低的情況下,有熒光信號的反應單元數目等于目標DNA分子的拷貝數。但是,在通常情況下,數字PCR的反應單元中可能包含兩個或兩個以上的目標分子,這時可以采用泊松概率分布公式( Poisson distribution) 進行計算。

上式中λ為每個反應單元中包含目標DNA分子的平均拷貝數(濃度),p為在一定的λ條件下,每個反應單元中包含 k 拷貝目標 DNA 分子的概率。λ由樣品溶液的稀釋系數 m 決定,有 λ= cm,其中c為樣品的原始拷貝數(濃度) 。當 k = 0 (不含目標 DNA 分子) 時,上式可簡化為 p = e^-λ = e^-cm, p 可以看作是無熒光信號的反應單元數與反應單元總數的比值,即

?
其中,n為反應單元總數,f為有熒光信號的反應單元數。上式兩邊取對數( ln) 得到
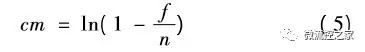
?
從數字 PCR 反應單元總數和有熒光信號的單元數以及樣品的稀釋系數,就可以得到樣本的最初拷貝數(濃度) 。數字PCR的定量方法不依賴于擴增曲線的循環閾值,因此不受擴增效率的影響,也不必采用看家基因( house-keeping gene)和標準曲線,具有很好的準確度和重現性,可以實現絕對定量分析。
數字 PCR 的靈敏度也可以稱之為分辨率,指的是對目標基因或突變的識別能力。它除了與檢測器的靈敏度和PCR擴增效率等因素有關外,很大程度上取決于反應單元的數目n。理論上每個反應單元至多有一個拷貝的 DNA 分子,相同體積的情況下, n = 10^2 時,該方法的最大分辨率為 1 /100,也就是說樣品濃度最低為1%可以被檢出。如果 n = 10^4 時,就可以從10^4個分子中檢測到1個靶標,即樣品濃度最低為 0. 01% 可以被檢出。因此,反應單元的數目越多,數字PCR的靈敏度越高,準確度也越高。
?
數字 PCR 技術提出至今,相關技術和產業化發展都非常迅速。迄今為止,數字 PCR 技術主要有三類: 微反應室/孔板、大規模集成微流控芯片和液滴數字PCR系統。
微反應室/孔板數字PCR
傳統 PCR 或定量 PCR 反應都是在96/384孔板中進行的,因此早期的數字PCR技術也采用 96 /384孔板作為反應單元。但是數字PCR技術的靈敏度取決于反應單元的總數n,因此,理論上反應單元數越多越有利于提高靈敏度和準確度,普通的96 /384孔板無法滿足檢測的需要。而且,在96 /384 孔板中進行的PCR反應體系通常大于5μl,由試劑消耗引起的高成本問題令人望而卻步。針對上述問題, Morrison 等在25mm×75mm不銹鋼芯片上刻蝕了3072個直徑為300μm的微反應室 (如圖2a所示),每個反應單元的體積降低至33 nl。該芯片可在商品化 PCR 儀上使用,與384 孔板的檢測靈敏度相當,但反應體積降低為原來的1/64,樣品通量提高了24倍。隨著反應單元數目的成倍增加,反應體積從微升級降至納升級,傳統的操作人員采用移液器加試樣的方式已經無法滿足快速精準取樣的要求,因此需要借助高通量自動點樣儀或機械手等設備,這無疑大幅提高了系統的成本和操作的復雜性。
大規模集成流路數字PCR
微流控芯片技術的發展為我們提供了一個實現低成本、小體積和高通量平行PCR分析的理想平臺。2000年,Unger等采用多層軟刻蝕 ( multilayer soft lithography,MSL) 技術在聚二甲基硅氧烷( polydimethylsiloxane,PDMS) 微流控芯片上設計并加工高密度微泵微閥結構(如圖2b所示) ,他們將這種芯片稱為IFC( integrated fluidic circuit)。IFC 利用PDMS材料具有高彈性的特點,通過多層軟刻蝕技術在芯片上加工交織的液體和氣體通道結構,可以快速并準確地將流體分成若干個獨立的單元,進行多步平行反應。2006 年Ottesen等將IFC芯片用于數字PCR分析,通過精準控制微泵微閥的開啟和關閉,一步操作即可將一個樣本平均分配到 1176個反應單元中,每個反應單元的體積只有6. 25 nl,成功代替了傳統點樣儀和384孔板。他們同時進行了6個樣本7 056個單元的平行數字PCR分析。此外, Hansen及其同事采用 MSL技術加工了具有10^6個結構單元的數字PCR 芯片,每個反應單元的體積降低至10 pl,芯片密度達到 440000 /cm2。與微反應室數字PCR系統相比,IFC的特點是通量更高,每個反應單元的體積更小,加樣更快。最近,Men等在2mm×2mm區域內加工了82000個 fl 級反應單元,進行數字PCR分析。
液滴數字PCR
液滴數字PCR源于乳液PCR( emulsion PCR) 技術,即將DNA模板與連接引物的磁性微球以極低的濃度(比如單拷貝) 包裹于油水兩相形成的納升至皮升級液滴中進行 PCR 擴增,擴增后的產物富集在磁性微球上,收集破乳后進行測序。通過油水兩相間隔得到的以液滴為單位的 PCR 反應體系,比微孔板和 IFC 系統更容易實現小體積和高通量,而且系統簡單,成本低,因此成為理想的數字PCR技術平臺。Vogelstein 及其同事提出的 BEAMing 技術就是一種基于乳液PCR的數字PCR系統。Lu 等也采用 BEAMing 和連接酶反應實現對 mRNA 的定量分析。Zhou 等在 BEAMing PCR 擴增后破乳,將連接不同產物的磁珠包被在聚丙烯酰胺凝膠中制成磁珠陣列進行熒光檢測。但是,上述技術需要將單拷貝 DNA 模板與磁珠同時包裹在一個液滴中,增加了系統的復雜性和定量分析的難度。Beer 等在微流控芯片通道中用油相包裹皮升級液滴,液滴中只包裹了單拷貝 DNA 模板、 PCR引物及試劑,實現了數字PCR 定量分析。Hindson 等在微流控芯片中生成了20000—2000000個體積為 1nl 的液滴,然后將液滴轉移到 96 孔板中進行TaqMan PCR擴增,至終點后將液滴從 96 孔板中取出,在微流控芯片中采用流式方法使液滴順次經過雙通道熒光檢測器,以1000個液滴/s的速度進行計數。Pekin 等設計了一種微流控芯片,可分別生成包含不同熒光探針和 DNA 模板的液滴,再進行液滴融合,他們將該系統用于KRAS基因突變分析。此外,Shen 等提出了一種通過滑動芯片( SlipChip) 液滴數字PCR系統,設計了帶有微流體通道和反應單元的玻璃芯片,上下兩片之間用油相密封,通過滑動將樣品溶液從流體通道中引入反應單元,同時生成1280個體積僅為2. 6 nl的液滴陣列,在芯片上進行PCR擴增和熒光成像分析。隨后,他們還設計了具有不同體積反應單元的 SlipChip,從1 nl變化到125 nl,僅用不到 200個反應單元,理論上可以實現12000個等體積反應單元所能達到的檢測濃度范圍。Yang 等還合成了凝膠微球作為反應單元進行數字 PCR 擴增和定量分析,與前述液滴相比凝膠微球體系更加穩定和可控,易于收集和保存擴增產物。
?
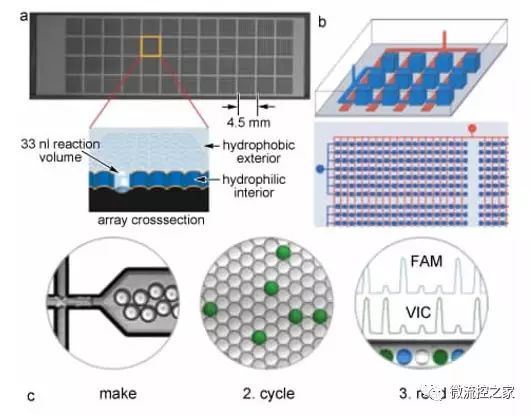
圖 2? 幾種典型數字 PCR 芯片: a) 微反應室/孔板數字 PCR( OpenArrayTM); b) 大規模集成微流控數字 PCR 芯片( BioMarkTM); c) 微液滴數字 PCR( QX100TM)
?
基因不穩定性分析
近年來隨著人類對癌癥的不斷研究和認識,大量證據表明癌癥是一種基因(染色體) 異常變化引起的疾病,普遍認可的異常情況包括癌基因及抑癌基因的突變、插入或缺失等。研究顯示,癌癥是由體細胞基因突變( somatic mutation),包括等位基因失衡( allelic imbalance,AI) ,雜合性缺失( loss of heterozygosity,LOH) ,拷貝數變異 ( copy number variations,CNVs) 等一系列基因變異情況引起的。 不過,癌細胞通常與大量正常細胞同時存在,因此,如何從大量正常細胞的 DNA 中檢測到少量的異常基因成為癌癥研究領域關注的焦點問題之一。Vogelstein及其同事提出數字 PCR 的初衷就是通過稀釋將樣品分配至盡可能多的單元中,從而將突變基因分子與大量的野生型基因分離開,在高背景條件下得到少量突變基因的信號。他們以KRAS基因突變為研究對象,對腸癌患者的糞便樣品進行了數字 PCR 分析,得到 KRAS 基因第12號密碼子點突變率約為4% (4/102)。接著,他們利用數字SNP技術研究了腸癌患者染色體18q雜合性缺失與血管侵入行為的關系,結果顯示13個沒有發生染色體 18q LOH的腫瘤樣本均沒有發現血管侵入行為,而 18個具有染色體18q LOH的腫瘤樣本中有17個發生了癌細胞的血管侵入。Shih等研究了卵巢漿液性腺癌( ovarian serous carcinoma) 患者腹水中BRAF基因 599位密碼子, KRAS 基因12和13位密碼子基因突變情況,及其他 7 種標志物在包括卵巢癌、胰腺癌和腸癌患者體液中的基因突變情況。 Hanlon 等則對多發性骨髓瘤( multiple myeloma) 病人骨髓活檢樣本中染色體 13q 14缺失情況進行了研究,不經任何細胞富集技術,他們在 18 個病人中檢測到16個LOH( 89% ) ,靈敏度高于傳統熒光原位雜交( FISH) 技術。Yung 等考察了35個非小細胞肺癌臨床血樣中表皮生長因子受體( EGFR) 基因外顯子 19 缺失及 L858R 突變情況,與直接測序技術相比,數字PCR技術在突變率低于30%的體液樣本中具有更高的靈敏度和檢出率。
人類基因組中存在一些范圍從kb到Mb片段的亞微觀結構變異,主要包括缺失、復制及嵌入等,統稱為拷貝數變異( CNVs)。近年來的研究發現, CNVs與包括 HIV-1 感染在內的多種疾病密切相關,對于研究基因變異在生物進化、生理過程及疾病進程等領域具有重要意義。現有的CNVs研究技術包括比較基因組芯片(microarray based comparative genomic hybridization,array-CGH) ,高密度寡核苷酸芯片( high density oligonucleotide array,FISH) ,測序及定量PCR等,但是上述技術在靈敏度和分辨率方面存在缺陷,例如測序只適用于高于 30%的變異率檢測,而定量PCR難以分辨低于2倍的CT差異,不足以滿足CNVs分析的要求。Ramakrishnan及其同事建立了一種用于研究CNVs的數字PCR模式及統計學方法,通過直接計數目標基因與參照基因(拷貝數為1的基因,例如RNase P)的數目,計算比值,得到目標基因的拷貝數情況,最高分辨率達到15% 。他們研究了40位乳腺癌病人和8位正常人組織樣本中HEBB2基因CNVs情況,其中14例癌癥組織樣本中HEBB2基因 CNVs 高于5。Wang 等對16個細胞系和20個腫瘤樣本中EGFR CNVs、外顯子19缺失及 L858R 突變情況進行了研究,他們在早期切除的肺癌腫瘤樣本中檢測到微量的對抗癌藥物敏感的EGFR突變( 0. 02%—9. 26% ),而且沒有發現EGFR基因的拷貝數變異情況。Hindson 等考察了人類基因組中MRGPRX1、染色體X和CYP2D6的CNVs情況,以及癌癥病人HEBB2基因CNVs。
-
焦點事件

-
焦點事件

-
綜述
-
焦點事件

